Microbial communities
Microbial communities underpin function of the planet. Ability to describe constituent members using 16S rDNA has become both a growth industry and a career opportunity. Especially when associated with the term “microbiome”. But left largely untouched is understanding of how it all works: the eco-evo dynamics of 1000s of interacting species. How does one progress beyond the descriptive?
One way to progress is to use metagenomic data as a way of accessing population genetic processes operating within communities. This is tough, but a recent paper gives hope. In understanding population genetic processes the goal is to say something about the likelihood that genes under selection fix. But what are the genes under selection? Even if we knew, would our understanding of population genetics apply to complex microbial communities? Which prompts one to ask of the evolutionary dynamics of a single genetic lineage within a community. I don’t know what it looks like, but I doubt that it will look like a “Darwin was right” curve. To be clear: Darwin was right.
Beyond these not inconsiderable challenges is the need to link genes to community function. This is where a sabbatical did a world of good. Along with a deliciously stinky compost heap in Square Théodore-Monod — known to be full of phages — and the adept experimental / computational skills of Steven Quistad.
Selfish genetic elements (SGEs) continually fascinate (see our work on REPINs and RAYTs, and integrative and conjugative elements (ICEs)). Even more so conditions leading to their extinction. In short, evolutionary death comes when SGEs cease to encounter new hosts. In the absence of new hosts (and thus lateral transfer) selection is powerless to eliminate mutant types with defects in transfer ability. The genomes of most organisms are littered with dead SGEs. In the bacterial world genome sequence reveals an abundance of phage-like elements that have lost ability to excise from the chromosome.
If failure to encounter new hosts castrates SGEs, then it follows that frequent opportunity to encounter new hosts will breath evolutionary life into SGEs.
One additional feature of SGEs that move between hosts is their ability to accidentally incorporate pieces of host DNA. Our hunch is that this is much more common than currently thought. It is certainly well known for phages known as transducing phages, and for ICEs and plasmids, but there is no reason to think that lytic phages and other SGEs might not also accidentally package and move host DNA.
If this is true (and perhaps the dynamic is much greater and more fluid than appreciated), and evolutionary life can be breathed into SGEs by frequent exposure to new hosts, then there is possibility to use SGEs to link genes to microbial community function via a simple experimental manipulation.
The strategy is best seen in context of the “compost experiment”. We took independent 1 g samples of compost comprising many hundreds of microbial genera and a whole zoo of eukaryotes to boot and used these to found mesocosms containing minimal M9 medium with a piece of paper as the sole carbon source. Once established, the communities in each mesocosm were divided in two and subject there after to either a “vertical” or “horizontal” transfer regime. The vertical regime was predicted to castrate SGEs, whereas the horizontal regime was predicted to fuel SGE evolution and facilitate lateral transfer of ecologically significant genes.
The vertical regime involved bi-weekly serial transfer of the contents of one mesocosm into a fresh bottle. The horizontal regime involved the same serial transfer protocol, but involved an additional step: at the time of transfer a sample of each horizontal mesocosm was collected, centrifuged (to eliminate bacterial cells) and the supernatant passed through a 0.22 µm filter. The filtrate — containing a cocktail of SGEs — from each horizontal mesocosm was then pooled and a sample of the mixture distributed across all horizontal bottle. The schema is shown below.
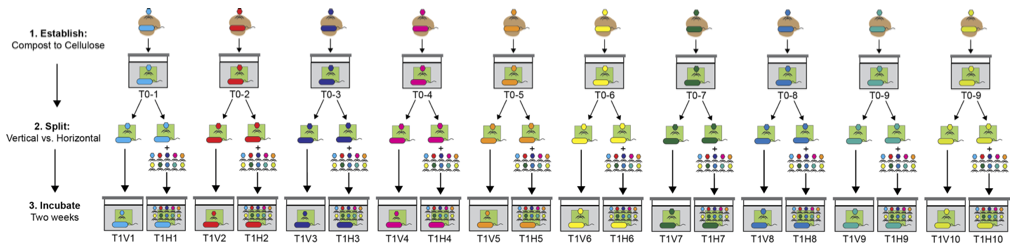
Two weeks after the split a metagenomic strategy was adopted to see whether SGEs and ecologically relevant genes were being mobilised. Because of the experimental design this was straightforward: sequence reads from the vertical bottle (e.g., T1V1) will be present in the T0-1 founding community. Sequence reads from the corresponding horizontal community (T1H1) ought to contain a set of reads not found in either the founding (T0-1) community or the vertical (T1V1) community. These reads (we refer to them as “unique” reads), if they exist, will be derived from another of the independent communities.
Did we find evidence of unique reads? Yes! Were there sufficient reads to assemble into contigs? Yes! Did the assembled contigs look like phages? Yes! Did they contain ecologically significant genes? Yes! Could we identify source communities? Yes!
Excited by these results we continued the experiment for another 48 weeks, implementing the horizontal / vertical transfer regimes at bi-weekly intervals. A pre-print is available here and contains the details. But in short, SGEs along with genes of ecological relevance were rapidly amplified across horizontal communities and we could even track their dynamics in communities composed of hundreds of microbial genera (there were even nematodes present in mesocosms after one year). In addition to genes implicated in degradation of cellulose (and particularly beta-glucosidases that define the rate-limiting step in cellulose degradation) there was a marked signal from genes implicated in nitrogen metabolism, and particularly ammonification. This told us that the mescosms were nitrogen-limited and prompted biochemical assays that showed a significant functional difference between communities subject to horizontal versus vertical treatments. Thus we have been able to demonstrate that via a simple and generally applicable manipulation that it is possible to link genes to community function.
Moving forward, Andy Farr is now applying this approach to study the environmental context of antibiotic resistance and we intend to extend the work further to incorporate nematodes and their microbiome. We would like to know much more about the SGEs involved (some appear to be entirely unknown) and we think also to incorporation of lineage selection regimes, where communities are subject to a death-birth process as units of selection in their own right. And then there are substrates — like poly(ethylene terephthalate) (PET) from which clothing and drink bottles are made — upon which communities might be selected and where what we do might even prove useful.